Advanced Skills¶
The previous tutorials should get you up and running so you can automate the construction of adsorbate geometries for a wide range of MOFs. However, for more advanced users, there are some additional features that may be of use. Let’s walk through some more complex examples.
Adding Multiple Adsorbates¶
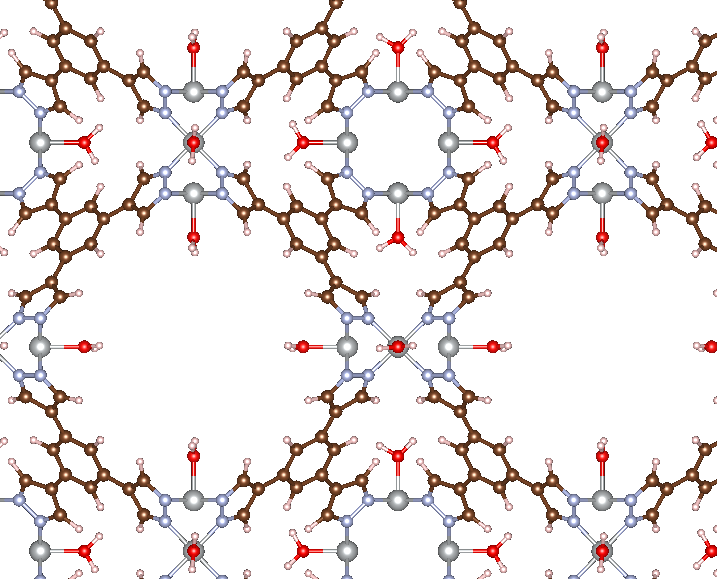
Instead of adding a single adsorbate, you may want to add an adsorbate to every metal site in a given MOF. We will consider the Ni-BTP MOF from the diatomic tutorial, except this time we will add an H2O molecule to every Ni site. This can be done using the code below.
import os
from mai.adsorbate_constructor import adsorbate_constructor
from ase.io import read, write
starting_mof_path = os.path.join('example_MOFs','Ni-BTP.cif') #path to CIF of MOF
#Get all Ni indices in ASE Atoms object of MOF
start_mof = read(starting_mof_path)
Ni_idx = [atom.index for atom in start_mof if atom.symbol == 'Ni']
#add H2O adsorbate
atoms = start_mof
for i, site_idx in enumerate(Ni_idx):
ads = adsorbate_constructor(ads='HOH',d_MX1=2.0,d_X1X2=0.96,d_X2X3=0.96,
ang_MX1X2=120,ang_triads=104.5,connect=2)
atoms = ads.get_adsorbate(atoms=atoms,site_idx=site_idx,write_file=False)
#Write out final CIF with all H2O molecules added
if not os.path.isdir('new_mofs'):
os.makedirs('new_mofs')
write(os.path.join('new_mofs','Ni-BTP_allH2O.cif'),atoms)
There are a few changes we’ve made to the usual workflow. The first is that we needed to identify all of the possible adsorption site indices, which we have defined as Ni_idx. This can be done using ASE’s built-in tools for working with ASE Atoms ojbects. We then iterate over each site and add an H2O molecule. Since MAI adds adsorbates sequentially, we don’t want to write out all the intermediate structures, so we also set write_file=False in get_adsorbate(). The last new aspect to introduce is that, instead of passing a CIF file via atoms_path in get_adsorbate(), we can directly pass in an ASE Atoms object via the atoms keyword argument. This is useful when you make modifications to the same MOF in a loop, as done here. The result of running this code is shown below.
Neighbor Algorithms¶
When specifying site_idx, the get_adsorbate() function will automatically use Pymatgen’s built-in nearest neighbor algorithms to determine the atoms in the first coordination sphere of the adsorption site. By default, MAI uses Pymatgen’s crystal algorithm, but additional algorithms are available as listed in get_NNs_pm() and described here. Through iterative testing, we have found crystal to be the best-performing algorithm for MOFs in general, but other algorithms can be considered if desired.
Pre-specification of Indices¶
In addition to the site_idx keyword in the get_adsorbate() function, it is also possible to manually specify the indices of the atoms in the first coordination sphere via the NN_indices keyword argument. This flexibility makes it possible to use a wide range of workflows in determining the desired adsorption site and coordinating atoms.