Potential Energy Grids¶
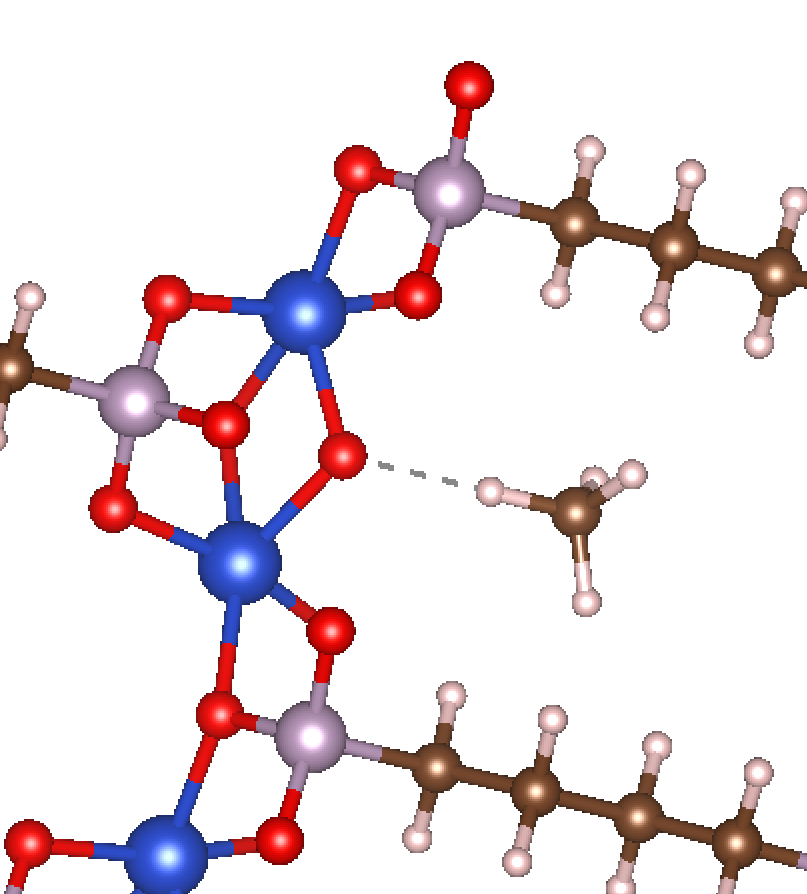
Some adsorbates do not lend themselves well to the geometric approaches laid out thus far. This is particularly the case for adsorbates that are physisorbed relatively far away from an adsorption site, as opposed to chemisorbed nearby. In these cases, an alternative way of initializing the adsorbate can be considered by mapping out a potential energy grid (PEG) of each MOF and putting the adsorbate in a low-energy site within some cutoff radius of the proposed adsorption site. A visualized example of such a PEG is shown below for a methane adsorbate near a proposed Ni-O active site.
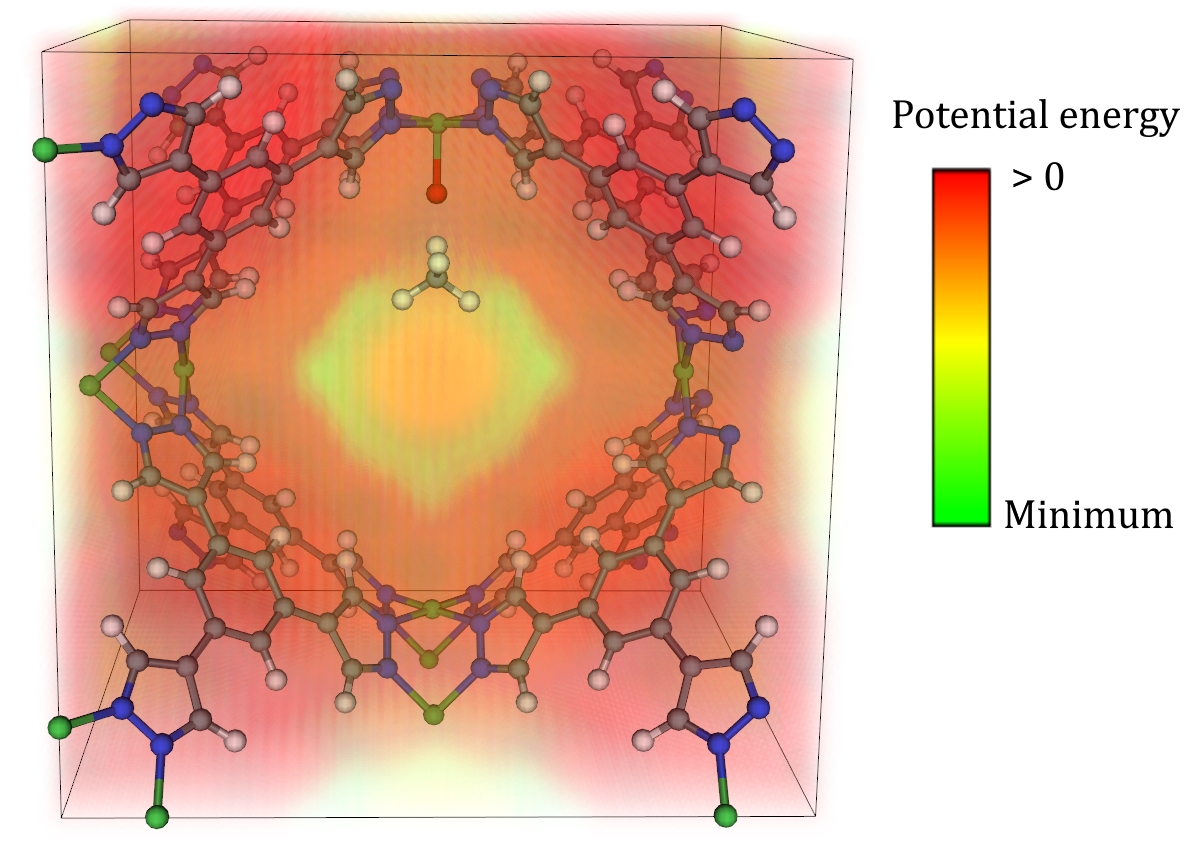
MAI supports two different formats for PEGs. The first is the cube file format, such as that generated from PorousMaterials.jl. Details of how to generate such PEGs can be found here. The second accepted format is a space-delimited file with four columns of (x,y,z,E) entries, where E is the potential energy and (x,y,z) are the coordinates. Each new line represents a new (x,y,z,E) vector. We’ll refer to this file as an ASCII grid.
Currently, only single-site CH4 adsorbates are supported with PEGs, although in principle it is trivial to consider other adsorbates as well. When using PEGs to initialize the position of CH4 adsorbates, the C atom of the CH4 molecule will be placed in the low-energy site, and the four remaining H atoms will be arranged to form the tetrahedral structure of CH4, with one of the H atoms pointed directly toward the adsorption site.
Unlike with the monatomics, diatomics, and triatomics tutorials, when dealing with PEGs, one must use the get_adsorbate_grid() function instead of get_adsorbate(). The get_adsorbate_grid() function is quite simple. An example code is shown below to initialize an adsorbate based on an example PEG and example MOF.
import os
from mai.adsorbate_constructor import adsorbate_constructor
from ase.io import read
grid_path = os.path.join('example_MOFs','energy_grids_ASCII') #path to PEG
mof_path = os.path.join(grid_path,'AHOKIR01-O.cif') #path to CIF of MOF
#Select the last O index in the MOF as the adsorption site
mof = read(mof_path)
site_idx = [atom.index for atom in mof if atom.symbol == 'O'][-1]
#Add the CH4 molecule to the O adsorption site within 3 A sphere
ads = adsorbate_constructor(ads='CH4',d_MX1=3.0)
mof_adsorbate = ads.get_adsorbate_grid(atoms_path=mof_path,
site_idx=site_idx,grid_path=grid_path,grid_format='ASCII')
The new arguments used in this code are described below:
- The
atoms_pathandsite_idxare the same as forget_adsorbate(). - The
grid_pathkeyword argument is the path to the PEG file. - The
grid_formatkeyword argument can be eithergrid_format='cube'orgrid_format='ASCII'.
The result of running the previous example code is the following structure.