Triatomics¶
In this example, we’ll work through how to add three-atom adsorbates to open metal sites in MOFs. The CIF for the MOF we’ll use for this example can be found here. This MOF is known as MIL-8BB and has trimetallic nodes with each metal cation in a square pyramidal geometry.
Contiguous Adsorbate¶
For this example, we will consider the initialization of a “contiguous” triatomic adsorbate. What I mean by this is that if we treat the adsorbate as an arbitrary molecule X1-X2-X3, then the adsorption process is described by M-X1-X2-X3. In this example, we’ll consider the adsorption of N2O to an Fe site, in both an η1-N and η1-O binding mode. The code to handle this is shown below.
import os
from mai.adsorbate_constructor import adsorbate_constructor
mof_path = os.path.join('example_MOFs','Fe-MIL-88B.cif') #path to CIF of MOF
#add N2O adsorbate in η1-N mode
ads = adsorbate_constructor(ads='N2O',d_MX1=2.0,d_X1X2=1.13,ang_MX1X2=180,d_X2X3=1.19)
new_mof_atoms1 = ads.get_adsorbate(atoms_path=mof_path,site_idx=0)
#add N2O adsorbate in η1-O mode
ads = adsorbate_constructor(ads='ON2',d_MX1=2.0,d_X1X2=1.19,ang_MX1X2=120,d_X2X3=1.13)
new_mof_atoms2 = ads.get_adsorbate(atoms_path=mof_path,site_idx=0)
Like with the previous examples, we need to initialize an adsorbate_constructor object and then provide it the MOF of interest. In the case of triatomics, we have a few new keywords to introduce. In addition to the arguments described in the previous examples, we can now provide MAI additional geometric parameters if desired. Namely, the new features are now that we can include the X2-X3 bond length and the X1-X2-X3 bond angle. The arguments used here are described below:
- The
ads,d_MX1X2,d_X1X2, andang_MX1X2are the same as before. - Now, we have the option to add the
d_X2X3keyword argument, which specifies the X2-X3 distance. It defaults tod_X2X3=d_X1X2if not specified. - We can also add the
ang_triadskeyword argument, which specifies the X1-X2-X3 bond angle. It defaults toang_triads=180if not specified.
That takes care of initializing the adsorbate_constructor object. With this, we provide the object with the path to the MOF, and it will initialize the adsorbate. Now let’s see what happens as a result of running this code! The initialized structures are shown below:
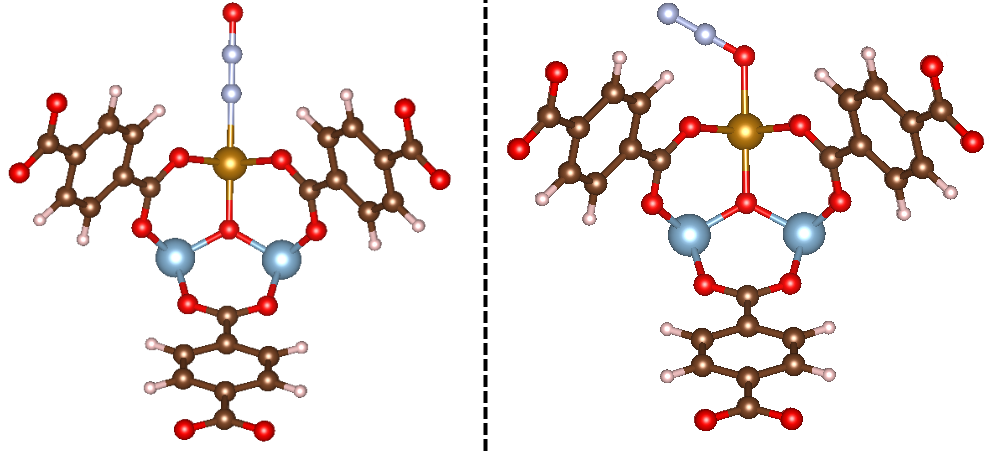
Exactly what we’d expect once more! You can see that in the first example, N2O is bound in an η1-N mode, whereas the second is bound in an η1-O mode, as specified. Feel free to play around with the bond distance and bond angle arguments to get a feel for how MAI works.
Noncontiguous Adsorbate¶
The last bit of trickery comes into play when dealing with what I’ll call “noncontiguous” adsorbates. These are adsorbates like water, where it is triatomic, but it is not bound in a sequential fashion. As with water, you will have a central atom of the adsorbate bound to the metal (instead of an M-O-H-H adsorption mode). To tell MAI about this desired adsorption mode, adsorbate_constructor has a keyword argument named connect, which is the atom number in ads that should be bound to the adsorption site. For the case of ads='HOH', we should set connect=2 to have the second atom (i.e. the O atom) bound to the adsorption site. Note that for this reason we cannot use ads='H2O' here.
An example code is shown below. The main thing to keep in mind is that now the connecting atom of the adsorbate is X2 instead of X1. If not specified, ang_triads will default to ang_triads=ang_MX1X2 if connect=2, but here we set it to 104.5°, as is standard for H2O.
import os
from mai.adsorbate_constructor import adsorbate_constructor
mof_path = os.path.join('example_MOFs','Fe-MIL-88B.cif') #path to CIF of MOF
#add H2O adsorbate
ads = adsorbate_constructor(ads='HOH',d_MX1=2.0,d_X1X2=0.96,d_X2X3=0.96,
ang_MX1X2=120.0,ang_triads=104.5,connect=2)
new_mof_atoms = ads.get_adsorbate(atoms_path=mof_path,site_idx=0)
This results in the following initialized structure:
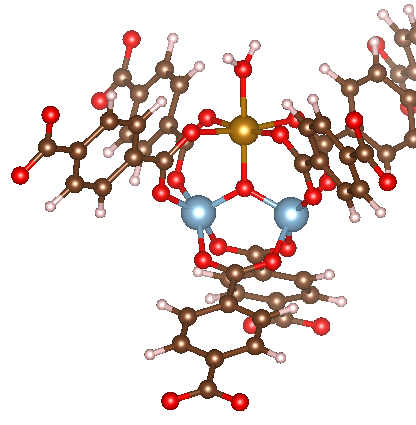
That concludes our tutorial for triatomic adsorbates.